








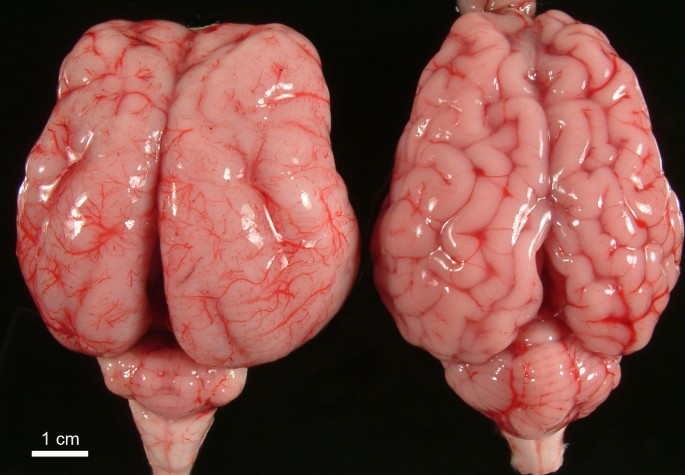
Lissencephalia typu 1 (angl. agyria, lissencephaly type I), tiež známa ako klasická lisencefália, je malformácia mozgu, ktorá sa môže vyskytnúť ako izolovaná abnormalita (izolovaná lisencefalická sekvencia [ILS]) alebo v spojení s určitými syndrómami (napr. Miller-Diekerov syndróm). Tento stav je charakterizovaný agýriou alebo pachygyriou, čo znamená absenciu alebo neúplný vývoj gyrifikácie mozgu alebo konvolúciu, čo spôsobuje, že povrch mozgu vyzerá nezvyčajne hladko, vyhladením mozgu.
Ak je prítomný základný syndróm, môžu existovať ďalšie príznaky a telesné nálezy. Môžu existovať rôzne možné príčiny izolovanej lisencefálie, vrátane vírusových infekcií, nedostatočného prekrvenia mozgu počas vývoja alebo určitých genetických faktorov. Na izolovanej lisencefálii sa podieľajú zmeny (mutácie) v niekoľkých génoch: LIS1, RELN, TUBA1A, NDE1, KATNB1, CDK5, ARX a DCX. Z nich boli najviac študované génové mutácie LIS1 a DCX.
Diagnostika:
Lisencefália môže byť dôsledkom rôznych negenetických (vonkajšie vplyvy) a genetických faktorov. Medzi tieto faktory môže patriť vnútromaternicová infekcia, prenatálna hypoxia mozgu (nedostatočný prísun okysličenej krvi do mozgu počas vývoja plodu) a/alebo rôzne génové mutácie.
Do izolovanej lissencefalie bolo zapojených niekoľko génových mutácií. Jedným z najlepšie študovaných príkladov je LIS1 alebo PAFAH1B1. Mutácie v tomto géne sú zodpovedné za lisencefáliu typu 1. Gén LIS1 je lokalizovaný na chromozóme 17p13.3. Tento gén kóduje izoformu 1B acetylhydrolázového faktora aktivujúceho doštičky, ktorá interaguje s proteínmi spojenými s mikrotubulami: dyneínom a dynaktínom. Táto interakcia je rozhodujúca pre správnu migráciu neurónov počas vývoja mozgu plodu; narušenie tejto interakcie má za následok lisencefáliu. Väčšina detí s izolovanou sekvenciou lissencefalie vykazuje mutácie alebo delécie iba génu LIS1, zatiaľ čo u dojčiat s Millerovým-Diekerovým syndrómom sa väčšinou zistilo, že majú mutácie v géne LIS1, ale majú tiež ďalšie delécie susedných génov na chromozóme 17, čo má za následok typ lissencefalie 1 rysy a iné kraniofaciálne abnormality. K takýmto chromozomálnym zmenám dochádza náhodne a sú pozorované iba u dieťaťa bez dôkazu o zmenách u oboch rodičov. Čo je dôležité, táto genetická forma lisencefalie sa v rodinách neopakuje, a preto je riziko ďalšieho dieťaťa s týmto ochorením extrémne nízke.
Z génov, ktoré sa podieľajú na lisencefalii, sú gény DCX a ARX pozoruhodné, pretože sú lokalizované na chromozóme X. Túto genetickú formu lissencefalie možno pozorovať u viac ako jedného dieťaťa v rodine, pretože mutácia môže byť prítomná v DNA zdravej matky. Lisencefália spôsobená DCX a ARX sa označuje ako lissencefália viazaná na X typu 1 a 2 (XLIS 1-2 alebo LISX 1-2). Pretože muži majú iba jeden X chromozóm, muži, ktorí zdedia gén choroby, majú väčšiu pravdepodobnosť manifestácie celého spektra abnormalít spojených s poruchou, a preto sú zvyčajne vážnejšie postihnutí. Ženy, ktoré zdedia túto génovú mutáciu, môžu mať variabilnejšiu podobu a môžu byť miernejšie postihnuté ako muži alebo môžu byť zdravé bez symptómov.
Gén DCX kóduje proteín doublecortin. Doublecortin sa spája s mikrotubulami na reguláciu migrácie neurónov. Mutácie spojené s X sa môžu objaviť náhodne alebo môžu byť dedičné. Gén ARX kóduje homeoboxový proteín súvisiaci s bez arististami. Okrem klasických znakov lissencefalie môžu deti s mutáciou ARX mať aj absenciu častí mozgu (hydranencefália), abnormálne genitálie, závažnú epilepsiu a ďalšie abnormality.
Ostatné génové mutácie, ktoré sú spojené s lissencefáliou, ako napríklad RELN, ktorá spôsobuje Norman-Robertsov syndróm, majú autozomálne recesívne dedičný model. Recesívne genetické poruchy sa vyskytujú vtedy, ak jedinec zdedí dve kópie abnormálneho génu pre rovnaký znak, jednu od každého rodiča. Ak jednotlivec zdedí jeden normálny gén a jeden gén pre chorobu, táto osoba bude nosičom choroby, ale zvyčajne nebude vykazovať príznaky. Riziko, že dvaja rodičia nosiča prejdú zmeneným génom a budú mať postihnuté dieťa, je 25% pri každom tehotenstve. Riziko mať dieťa, ktoré je nosičom ako rodičia, je pri každom tehotenstve 50%. Šanca, že dieťa dostane normálne gény od oboch rodičov, je 25%. Riziko je rovnaké pre mužov a ženy.
Okrem LIS1, RELN, DCX a ARX bolo zistené, že lisencefáliu spôsobujú aj mutácie v iných génoch. Patria sem: TUBA1A, NDE1, KATNB1 a CDK5. Tieto gény zdieľajú molekulárnu funkciu s LIS1 a DCX, ktoré fungujú ako súčasť bunkového aparátu dyneínu a dynaktínu potrebného na migráciu neurónov počas vývoja mozgu plodu.
Objavujúce sa dôkazy naznačujú, že genetické zmeny a negenetické príčiny vedú k lisencefálii v dôsledku zhoršenej migrácie neurónov vonkajšej oblasti mozgu počas vývoja plodu. Mozgová kôra, ktorá je zodpovedná za vedomý pohyb a myslenie, sa bežne skladá z niekoľkých hlbokých gyrov a sulcov (výbežok - brázd), ktoré sú tvorené „skladaním“ mozgovej kôry. Počas embryonálneho rastu novovytvorené bunky, z ktorých sa neskôr vyvinú špecializované nervové bunky, normálne migrujú na povrch mozgu (migrácia neurónov), čo vedie k tvorbe niekoľkých bunkových vrstiev. V prípade lisencefalie typu 1 však bunky nedokážu migrovať na určené miesto, čo má za následok neuronálnu dysmigráciu, a v mozgovej kôre sa vyvinie nedostatočný počet bunkových vrstiev s absenciou alebo neúplným rozvojom gyrov.
Celkový výskyt lisencefálie je zriedkavý a odhaduje sa okolo 1,2 na 100 000 živonarodených detí.
Lisencefáliu typu 1 je možné diagnostikovať dôkladným klinickým vyšetrením, zobrazovacími metódami mozgu vrátane USG mozgu (u novorodencov cez veľkú fontanelu), pomocov skenov počítačovou tomografiou (CT mozgu) a/alebo zobrazovaním magnetickou rezonanciou (MRI mozgu) a genetickým testovaním, ako je chromozomálna analýza a/alebo špecifická analýza génovej mutácie. Počas CT skenovania sa počítač a röntgenové lúče použijú na vytvorenie filmu, ktorý zobrazuje prierezové obrazy štruktúry mozgového tkaniva. Pri magnetickej rezonancii magnetické pole a rádiové vlny vytvárajú prierezové obrazy mozgu. Ďalšímí vyšetrovacími metódami, ktoré môžu dopomôcť k diagnostike, je elektroencefalogram (EEG mozgu). Počas EEG sa zaznamenávajú elektrické impulzy mozgu. Malformácie mozgu, vrátane lisencefálie, sú často spojené s abnormálnymi elektrickými impulzmi mozgu a/alebo záchvatmi. Abnormálny vzor EEG môže upriamiť pohľad a viesť k diagnostike lisencefálie. Nakoniec, DNA analýza môže detegovať určité delécie/mutácie v génoch spojených s lisencefáliou. Teraz je k dispozícii komerčne dostupné testovanie génov na známe genetické príčiny lisencefálie. Počet génov zahrnutých v týchto testoch sa stále zvyšuje s ďalším výskumom.
Terapia:
Je zameraná na špecifické symptómy, ktoré sú typické u každého jednotlivca. Liečba môže vyžadovať koordinované úsilie tímu špecialistov. Pediatri, neurológovia a ďalší zdravotnícki pracovníci môžu potrebovať systematicky a komplexne naplánovať liečbu dieťaťa s lisencefáliou.
Odporúčaná je rehabilitácia pre oneskorený psychomotorický vývoj, logopedické sedenia pre poruchu príjmu potravy alebo problémy s kŕmením, metódy stimulujúce intelektuálnu stránku dieťaťa (podporné terapie - animoterapia, ergoterapia, špeciálno-pedagogické sedenia, etc). Korekcia poruchy zraku a sluchu v prípade potreby.
Terapie pre jednotlivcov s lisencefáliou typu 1 sú symptomatické a podporné. Liečba môže zahŕňať opatrenia na zlepšenie príjmu živín u dojčiat s ťažkosťami s kŕmením; podávanie antikonvulzívnych liekov na prevenciu alebo redukciu kŕčových stavov (antiepileptiká).
Rodinám detí s lisencefáliou sa odporúča genetické poradenstvo.
Prejavy:
- mikrocefália (malý obvod hlavičky u detí)
- prítomnosť kŕčových stavov
- porucha intelektu až ťažké mentálne postihnutie
- neprospievanie, problémy s kŕmením
- porucha rastu - spomalený rast
- zhoršené motorické schopnosti
- štrukturálne abnormality mozgu - neprítomnosť alebo nevyvinutý corpus callosum (spleť nervov, ktorá spája prevú a ľavú hemisféru)
- typické črty tváre - malá brada (mikrognácia), vystúpené spánkové výbežky
- hypotónia v prvých rokoch života, neskôr až hypertonus
Na mape sú vyznačené rodiny so syndrómom - Lisencefália
Po registrácii môžete tieto rodiny kontaktovať. Prípadne Vás v budúcnosti môže kontaktovať druhá rodina, s ktorou si môžete vymeniť informácie.
Ak máte tento syndróm, zaregistrujte sa