








Pelizaeusova-Merzbacherová choroba
PMD syndróm, HLD1 syndróm, Hypomyelinizacia leukodystrofia 1
Popis:
Pelizaeusova-Merzbacherová choroba (angl. Pelizaeus-Merzbacher disease, PMD, hypomyelinating leukodystrophy 1, HLD1, sclerosis, diffuse familial brain) patrí medzi dedičné metabolické poruchy spôsobené abnormálnym metabolizmom myelínu - tzv. leukodystrofie. Ide o chorobu viazanú na pohlavný chromozóm X - tým pádom sú typicky postihovaní predovšetkým muži. Stupeň postihnutia má veľkú variabilitu a v priebehu života sa zjavne mení. V popredí stojí postihnutie centrálneho nervového systému, napríklad abnormálne pohyby očných bulbov, postihnutie psychomotorického vývoja, ataxia a niektoré poruchy hybnosti. Do dnešného dňa nevieme Pelizaeusovu-Merzbačovu chorobu kauzálne liečiť - v popredí je iba podporná liečba, napríklad intenzívna rehabilitácia.
Leukodystrofia patrí do veľmi širokého spektra genetických porúch (DMP), teda chorôb, pri ktorých dochádza k narušeniu vybraných metabolických dráh a dochádza k nerovnováhe v tvorbe alebo na rozklade určitých látok. Konkrétne u leukodystrofií je abnormálny metabolizmus myelínu, čo je kľúčová súčasť tzv. bielej hmoty v centrálnom nervovom systéme (CNS). Leukodystrofia sa prejavuje rozličnými neurologickými problémami (ktoré majú pôvod v poškodení CNS), napríklad oneskorením alebo regresiou psychického vývoja, poruchami motoriky alebo potrebou rozvoja kŕčnových stavov. Medzi leukodystrofie patria napríklad X-viazaná adrenoleukodystrofia, Krabbeho globoidná leukodystrofia, metachromatická leukodystrofia a Pelizaeusova-Merzbacherová choroba.
Pelizaeusova-Merzbacherová choroba bola prvýkrát popísaná v Nemecku neurológom Friedrichom Christophom Pelizaeusom v roku 1885 a neskôr aj nezávisle od nemeckého neuropatológa a psychiatra Ludwiga Merzbachera v roku 1910. Príčina tejto choroby bola zistená až oveľa neskôr v 20. storočí. Kauzálny gén pre toto ochorenie je lokalizovaný na dlhom ramienku pohlavného chromozómu X (Xq22.2) a jeho označenie je PLP1 (gén proteolipidového proteínu 1). Dnes už vieme, že pod klinickým pojmom Pelizaeusova-Merzbacherova choroba sa môžu zahrnúť viaceré rôzne choroby (spôsobené defektmi rôznych génov), ktoré sú všeobecne známe ako hypomyelinizujúce leukodystrofie (napríklad SPG2, PMLD1, SPG44, GJC2). Keďže je táto choroba viazaná na pohlavný chromozóm X - väčšinu postihnutých tvoria muži (chlapci), pretože majú za iba jeden chromozóm X (karyotyp 46, XY); ženy (s karyotypom 46, XX) vystupujú ako "prenášačky". Zodpovedný gén PLP1 má 7 exónov, kódujúcich proteín v celkovej dĺžke 277 aminokyselín so 4 transmembránovými doménami. Je exprimovaný najmä v oligodendrocytoch (gliové bunky v CNS). Okrem mutácií v PLP1 sa môže patologicky prejaviť aj jeho duplikácia alebo dokonca triplikácia.
Klinické prejavy Pelizaeus-Merzbacherej choroby sú veľmi variabilné. Klasická forma choroby trvá dlhé roky, niektoré príznaky môžu byť rozpoznané už v relatívne mladom veku. V prvých rokoch života sa prejavuje Pelizaeus - Merzbachova choroba ako svalová hypotónia a nystagmus (rytmický kmitavý pohyb očných bulb), oneskorený psychomotorický vývoj. Po nejakom období môže nystagmus zmiznúť, alebo sa zlepšiť, avšak začnú prevládať iné prejavy choroby ako spasticita - svalová stuhnutosť, neurologické prejavy charakteru ataxie (zlá koordinácia pohybov) alebo rôznych choreiformných pohybov (mimovoľné zášklby), dochádza k narušeniu kognitívneho vývoja, objavuje sa výrazná muskulárna hypotónia a mimo neurologických príznakov sa môže spomenúť aj skolióza alebo laryngeálny stridor (hlasné nadychovanie až pískanie). Reč zostáva zachovaná. U detí býva typický útlejší vzhľad a slabé prospievanie. Pri vrodenej forme začína ochorenie hneď po narodení, kedy sa postihnuté deti nenaučia chodiť a horné končatiny nedokážu naplno ovládať. V tomto prípade môže byť zhoršená aj verbálna komunikácia.
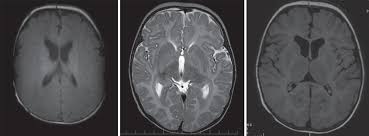
Diagnostika:
Pre diagnostiku choroby je dôležité neurologické vyšetrenie vrátane zobrazovacieho vyšetrenia CNS - typickou známkou je hypomelinizácia pri vyšetrení MRI mozgu. Diagnóza v rodine potvrdí molekulárne genetické vyšetrenie.
Liečba:
Pelizaeusova-Merzbacherová choroba je bez známej kauzálnej liečby. Postihnutému môže pomôcť len symptomatická liečba - napríklad intenzívna rehabilitácia, včasné stimuly alebo chirurgické zákroky na riešenie stridoru alebo skoliózy. V rodinách s výskytom Pelizaeusovych-Merzbacherových chorôb je možná (v prípade identifikácie génového defektu) cielená prenatálna diagnostika. Pre dysfágiu (nemožnosť prehĺtania) je možnosťou prijímu potravy cez PEG - sondou zavedenou do žalúdka. Epileptické a kŕčovité stavy sa liečia antiepileptikami
Prejavy:
- abnormálne okohybné pohyby (nystagmus)
- ataxia (nesprávna koordinácia pohybov)
- choreiformné pohyby (mimovoľné zášklby)
- narušenie duševného vývoja
- svalová hypotónia až spasticita svalov
- skolióza
- laryngeálny stridor
- kŕčovité záchvaty
- psychomotorické oneskorenie, neprospievanie
Na mape sú vyznačené rodiny so syndrómom - Pelizaeusova-Merzbacherová choroba
Po registrácii môžete tieto rodiny kontaktovať. Prípadne Vás v budúcnosti môže kontaktovať druhá rodina, s ktorou si môžete vymeniť informácie.
Ak máte tento syndróm, zaregistrujte sa- PMD Foundation
- National Organization for Rare Diseases
- United Leukodystrophy Foundation
- CLIMB (Children Living with Inherited Metabolic Diseases)
- ELA – European Association Against Leukodystrophies
- PMD Family Support - podporná skupina pre rodiny