








Vrodená metabolická porucha
Dedičná metabolická porucha
Popis:
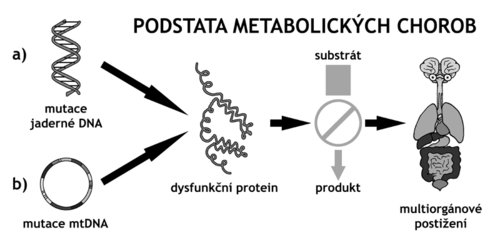
Vrodené metabolické poruchy (správnejšie dedičné metabolické poruchy (DMP) predstavujú heterogénnu skupinu približne 800-900 genetických ochorení, ktorých spoločným znakom je prítomnosť biochemických alebo enzymatických výnimiek, ktoré je možné zistiť iba špeciálnym vyšetrením. DMP sú typickými predstaviteľmi skupiny vzácnych chorôb ( "rare diseases"). Sú dedené väčšinou autozomálne recesívnym či gonosomálne recesívnym, ale aj dominantným spôsobom, u niektorých je dedičnosť mitochondriálna. Väčšina dedičných metabolických porúch je spôsobená poškodením génu, ktorý kóduje syntézu enzýmu uľahčujúceho premenu rôznych látok (substrátov) na látky iné (produkty). Zdravotné ťažkosti pacientov s týmito ochoreniami vznikajú buď v dôsledku hromadenia látok, ktoré sú pre organizmus toxické alebo narušujú jeho normálne funkcie, alebo kvôli zníženej schopnosti syntetizovať pre organizmus nevyhnutné zlúčeniny.
Počiatky objavovania dedičných metabolických porúch sú spojené s menom Archibald Garrod, ktorý ako prvý poukázal na súvislosť ľudských chorôb a Mendelových zákonov dedičnosti a formuloval koncept dedičných metabolických porúch (inborn errors of metabolism). Garrod sa zaoberal štúdiom alkaptonúrie a v roku 1902 publikoval knihu The Incidencia of alkaptonury: a Study in Chemical Individuality, ktorá je prvým záznamom prípadu recesívnej dedičnosti u ľudí. V roku 1923 vyšla ďalšia jeho kniha s názvom Inborn Errors of Metabolism, v ktorej publikoval svoje štúdie o alkaptonúrii, cystinurii, pentosurii a albinismu. Práve prekladom termínu inborn errors of metabolism vznikol u nás dlho používaný nepresný názov vrodené metabolické poruchy (vrodené metabolické chyby).
Príčinou DMP je geneticky podmienená porucha funkcie enzýmu či transportného proteínu. Molekulovou podstatou tejto dysfunkcie je zvyčajne homozygocia či zmiešaná heterozygocia u chorôb s autozomálne recesívnym spôsobom prenosu, hemizygocia u chorôb s gonozomálne recesívnym prenosom a prítomnosť mutácií v nadkritickom množstve organel u chorôb s mitochondriálnou dedičnosťou. U časti DMP sú mutácie v príslušných génoch jediným postačujúcim faktorom pre rozvoj klinicky viditeľnej choroby. Naproti tomu u ostatných DMP je pre klinickú manifestáciu (okrem génovej mutácie) potrebné vystavenie pacienta látke, ktorú nedokáže spracovávať.
V súčasnej dobe už nie je možné vychádzať z predstavy, že výskyt dedičných metabolických ochorení v populácii je vzácny a že sa s nimi praktický lekár počas svojej praxe nestretne. Diagnóza metabolických ochorení vyžaduje úzku spoluprácu medzi praktickými lekármi a špecializovanými laboratóriami.
Z hľadiska metabolitov, ktoré spôsobujú klinické prejavy ochorenia, možno dedičné metabolické poruchy rozdeliť na choroby malých molekúl a choroby veľkých (komplexných) molekúl. V nasledujúcom texte bude použitá klasifikácia definovaná metabolickou dráhou alebo typom metabolitu.
Klinické príznaky metabolických ochorení sú rôznorodé a môžu sa objaviť u detí aj v neskoršom veku. Ich závažnosť závisí od typu molekulárneho postihnutia, stupňa enzýmového deficitu a funkciu postihnutého enzýmu alebo iného proteínu v metabolických dejoch. V novorodeneckom a ranom dojčenskom veku sa manifestujú závažné deficity kľúčových enzýmov a priebeh týchto ochorení je často smrteľný.
Niektoré ochorenia sa môžu prejaviť príznakmi špecifickými (typickými pre dané ochorenie), inokedy sa metabolická porucha prejaví nešpecificky (príznaky, ktoré nachádzame aj u iných ochorení).
Medzi špecifické príznaky patria napríklad ektópia šošovky a tromboembolické príhody u homocystinúrie.
K nešpecifickým príznakom sa radia poruchy psychomotorického vývoja, hypotónia, nechutenstvo, neprospevanie. Až postupne dochádza k rozvoju postihnutiu funkcie jednotlivých tkanív (hypertrofická kardiomyopatia, demyelinizačné postihnutia CNS, hepatomegália, hepatopatia, katarakta, obličková nedostatočnosť, atď.).
Kombináciou nešpecifických príznakov niekedy vzniká typický klinický obraz. Napríklad u novorodenca s výraznou hypotóniou (zníženie svalového napätia), klenutým čelom, širokým koreňom nosa, zväčšením pečene a sleziny, poškodením pečene a polycystickými obličkami možno usudzovať o peroxizomálnych ochoreniach.
Podozrenie na prítomnosť metabolickej poruchy by mali vzbudiť najmä tieto príznaky:
- nevysvetliteľná psychomotorická retardácia, poruchy svalového napätia, kŕče
- neobvyklý zápach potu a moču
- opakované epizódy nejasného vracanie, acidóza, zmeny správania, poruchy vedomia
- hepatomegália
- obličkové kamene
- anamnéza o konsanguinite, opakovaných spontánnych potratoch, nevyjasnených úmrtiach pod obrazom "sepsy", výskyte SIDS alebo atak Reye-like syndrómu v rodine, by mali vždy viesť k podozreniu na dedičné metabolické ochorenie
Podľa rýchlosti nástupu klinických príznakov rozlišujeme metabolické ochorenia s akútnym, intermitentným a chronickým priebehom.
Akútne metabolické ochorenia sa zvyčajne začínajú prejavovať už v novorodeneckom alebo ranom dojčenskom veku, aj keď existujú aj neskoré formy týchto ochorení. Často možno vysledovať provokujúci spúšťací mechanizmus rozvoja klinických príznakov. U porúch metabolizmu aminokyselín, porúch v cykle močoviny dochádza k prejavom metabolického ochorenia po začatí perorálnej výživy, pri prechode na stravu s vyšším obsahom bielkovín alebo v priebehu katabolizmu pri infekciách. U porúch beta-oxidácie mastných kyselín je to hladovanie alebo nedostatočné krytie zvýšených energetických nárokov organizmu pri záťaži (interkurentná infekcia, stres, fyzická námaha, očkovanie). Pri každom donosenom fyziologickom novorodencovi alebo malom dojčati, u ktorého dôjde po bezpríznakovom období k prudkému zhoršeniu klinického stavu, keď dieťa prestáva piť, zvracia, objavuje sa porucha vedomia, kŕče alebo respiračné zlyhanie, by malo byť vždy v rámci diferenciálne diagnostických úvah pomyslenie na dedičné metabolické ochorenie. Rad týchto ochorení je liečebne ovplyvniteľná včasným začatím terapeutických postupov, medzi ktoré patrí napríklad eliminácia toxického metabolitu z organizmu pomocou hemodiafiltrácií, diétne opatrenia (nízkobielkovinová diéta, vylúčenie niektorých zložiek zo stravy) alebo režimové opatrenia (antihypoglykemický režim).
Metabolické ochorenia s intermitentným priebehom sa manifestuje v atakoch, vyprovokovaných zmenou výživy alebo zvýšenou energetickou potrebou organizmu v priebehu akútnych stavov vyvolávajúcich katabolizmus. Medzi atakmi bývajú chorí bez akýchkoľvek klinických ťažkostí a tiež stanovenie diagnózy je viazané na obdobie dekompenzácie ochorenia (napríklad prechodná forma leucinózy, opakované ataky Reye-like syndrómu u niektorých porúch beta-oxidácie mastných kyselín, neskoré formy deficitu ornitintranskarbamylázy - porucha v cykle močoviny, deficit fruktózovo-1,6-bisfosfatázy z porúch glukoneogenézy).
Najčastejším prejavom chronicky progresívneho metabolického ochorenia je postupné spomalenie, zástava či dokonca regres pôvodne normálneho psychomotorického vývoja, nastupujúci po rôzne dlhom bezpríznakovom období. Takto sa začínajú manifestovať lysozomálne ochorenia zo skupiny mukopolysacharidóz a glykoproteinóz, u ktorých sa vyvíja faciálna dysmorfia s hrubými rysmi v tvári a prejavy orgánového ukladania (hepatosplenomegália, zákaly rohoviek, dysostosis multiplex, chlopňové chyby, rozvoj hypertrofickej kardiomyopatie). Neurodegeneratívne metabolické ochorenia majú v klinickom obraze okrem deteriorácie psychomotorického vývoja progredujúcu neurologickú symptomatológiu (Krabbeová choroba, metachromatická leukodystrofia, X-viazaná adrenoleukodystrofia, gangliosidózy, neurónový ceroidlipofuscinózy, Lesh-Nyhanov syndróm). Poruchy mitochondriálneho energetického metabolizmu sa prejavuje najmä postihnutím funkcie energeticky náročných tkanív (CNS, srdce, svaly, pečeň) a všeobecnými príznakmi (neprospievanie, porucha rastu).
Diagnostika:
V prípade dedičnej metabolickéj poruchy spôsobenej deficitom enzýmu alebo transportného proteínu dochádza v mieste metabolického bloku k hromadeniu špecifických metabolitov. Podozrenie na určitú DMP možno potom vysloviť buď na základe stanovenia zvýšenej koncentrácie metabolitov hromadiacich sa nad metabolickým blokom alebo stanovením zníženej koncentrácie (event. neprítomnosti) metabolitov pod týmto blokom. Podozrenie na diagnózu DMP potom musí byť overená, čo je možné vykonať na úrovni enzýmu alebo génu.
Liečba:
Moderná liečba pacientov s DMP je založená na znalosti patogenézy chorôb a závisí od typu ochorenia a jeho klinickej závažnosti. V súčasnej dobe je známa liečba približne u jednej tretiny pacientov s DMP. V akútnej fáze ochorenia sa používa hemodialýza alebo hemodiafiltrácia na odstránenie toxických metabolitov z organizmu. Pre udržanie dlhodobej metabolickej kompenzácie u porúch metabolizmu aminokyselín sa používa nízkobielkovinová diéta doplnená o esenciálne (nepostrádateľné) aminokyseliny prostredníctvom špeciálneho diétneho prípravku. U porúch beta-oxidácie mastných kyselín sa odporúča častá strava s obmedzeným obsahom tukov. V liečbe niektorých DMP sa uplatňuje podávanie vysokých dávok vybraných vitamínov. U časti pacientov s lysozomálnym ochorením je účinné injekčné podávanie rekombinantného enzýmu, u iných pacientov s týmto typom ochorenia sa uplatňuje transplantácia hematopoetických kmeňových bunkách alebo orgánové transplantácie (pečene, obličiek, srdca).
Primárna prevencia DMP nie je s ohľadom na ich genetický pôvod možná. Veľký význam však má prevencia sekundárnej, ktorá spočíva vo včasnej diagnostike konkrétnej DMP. Klasickým príkladom úspešnej sekundárnej prevencie ochorení sa včasne začatú príslušnou liečbou (ak je liečiteľné) sú novorodeneckej skríningové programy. S výnimkou chorôb s mitochondriálnej dedičnosťou je úplná väčšina DMP diagnostikovateľná v rizikových rodinách prenatálnym vyšetrením.
Prejavy:
- porucha funkcie enzýmu
- porucha funkcie transportného proteínu
- rôznorodosť
- môžu sa objaviť aj u detí v neskoršom veku
- spomalený psychomotorický vývin
Typy vrodených metabolických porúch:
- Poruchy metabolizmu jednoduchých sacharidov
- Poruchy metabolizmu aminokyselín - fenylketonúria atď.
- Poruchy metabolizmu mastných kyselín
- Poruchy intermediárneho metabolizmu
- Klasické organické acidúrie (organic acidurias) - napr. Alcaptonuria
- Mitochondriálne choroby, napr. Kearnsov-Sayrov syndróm
- Poruchy metabolizmu purínov a pyrimidínov - Lesch-Nyhanov syndróm
- Poruchy metabolizmu porfyrínov
- Poruchy metabolizmu komplexných sacharidov polysacharidov (glykoproteíny, proteoglykány, mukopolysacharidy, oligosacharidy)
- Poruchy metabolizmu komplexných lipidov (sfingolipidy, mukolipidy)
- Poruchy metabolizmu lipoproteínov
- Poruchy metabolizmu steroidov
- Poruchy metabolizmu peroxizómov
Na mape sú vyznačené rodiny so syndrómom - Vrodená metabolická porucha
Po registrácii môžete tieto rodiny kontaktovať. Prípadne Vás v budúcnosti môže kontaktovať druhá rodina, s ktorou si môžete vymeniť informácie.
Ak máte tento syndróm, zaregistrujte sa